Культура клітин
Клітинні лінії раку підшлункової залози AsPC-1 (Cat# SCSP-5080), Capan-2 (Cat# SCSP-5310), Mia-PaCa-2 (Cat# TCHu271) і SW1990 (Cat# TCHu201) були отримані з банку клітин Китайської академії наук. PANC0203 (Cat# CRL-2553), PANC1005 (Cat# CRL-2547), Panc0327 (Cat# CRL-2549), BXPC3 (Cat# CRL-1687), Capan-1 (Cat# HTB-79) були отримані з Американської колекції типових культур (ATCC). HuP-T4 (кат. № CBP60540) було отримано від Cobioer. Ці клітинні лінії культивували в рекомендованих умовах. Усі клітинні лінії зберігали у зволоженому інкубаторі при 37 °C з 5% CO2. BXPC3 і Capan-2 культивували в середовищі RPMI1640 (Bioagrio), доповненому 10% фетальної бичачої сироватки (FBS, Vazyme). Capan-1 культивували в середовищі Iscove, модифікованому Дульбекко (IMDM, Servicebio), доповненому 20% FBS. PANC0203, PANC1005 і Panc0327 культивували в RPMI1640 з 15% фетальної бичачої сироватки та 1‰ інсуліну. Mia-PaCa-2 підтримували в модифікованому середовищі Ігла Дульбекко (DMEM) (Bioagrio), доповненому 10% FBS і 2,5% кінської сироватки (Beyotime). SW1990 і AsPC-1 культивували в DMEM з додаванням 10% FBS. HuP-T4 культивували в модифікованому середовищі Ігла (MEM, Bioagrio) з 20% FBS і 1% незамінних амінокислот (Thermo Fisher) з 1 мМ пірувату натрію (Thermo Fisher).
У все культуральне середовище додавали пеніцилін-стрептоміцин (P/S, Bioagrio). Усі клітинні лінії регулярно перевіряли на контамінацію мікоплазмою за допомогою набору швидкого виявлення мікоплазми Quick Cell та аутентифікували за допомогою профілювання коротких тандемних повторів (STR), щоб підтвердити їхню ідентичність. Клітини пасували при 70-80% конфлюентності з використанням 0,25% трипсину-EDTA (Gibco).
Екстракція РНК, побудова бібліотеки та секвенування РНК з довгим зчитуванням нанопор
Із зразків екстрагували загальну РНК, і полі(А) + мРНК виділяли за допомогою модуля магнітної ізоляції мРНК NEBNext Poly(A) (New England BioLabs), дотримуючись протоколу виробника для вибіркового збагачення полі(А) РНК. Цей метод забезпечив видалення рРНК та більшості інших некодуючих видів РНК, таким чином забезпечуючи високоякісну мРНК для подальших застосувань. Для довгострокового секвенування РНК синтез кДНК проводили за допомогою протоколу перемикання ланцюгів, наданого Oxford Nanopore Technologies (ONT). У цьому протоколі синтез першого ланцюга включає унікальну послідовність на 3'-кінці кДНК, щоб отримати інформацію, специфічну для ланцюга. Повнорозмірні кДНК-бібліотеки потім були підготовлені з полі(А)-відібраної мРНК за допомогою ONT кДНК-ПЛР-набору для секвенування (SQK-PCS109). кДНК ампліфікували за допомогою ПЛР протягом 13-14 циклів, використовуючи спеціальні адаптери зі штрих-кодом із набору Oxford Nanopore PCR Barcoding Kit (SQKPBK004) для полегшення мультиплексування зразків. Після ПЛР-ампліфікації 1D-адаптер для секвенування лігували з кДНК, щоб підготувати зразки для секвенування. Ці підготовлені бібліотеки згодом завантажували в проточну кювету FLOPRO002 R9.4.1, яку потім вставляли в секвенатор PromethION для високопродуктивного секвенування. Процесом секвенування керували за допомогою MinKNOW (версія 23.07.12), власного програмного забезпечення ОНТ, яке дозволяло в режимі реального часу відстежувати продуктивність циклу та контролювати якість. Базовий виклик проводився за допомогою Dorado (версія 7.1.4, https://github.com/nanoporetech/dorado) з використанням моделі високої точності. Усе секвенування проводилося компанією Wuhan Benagen Technology Co., Ltd. (Ухань, Китай), забезпечуючи високоякісний вихід для подальшого аналізу.
Обробка даних тривалого зчитування RNA-seq
Файли FASTQ тривалого зчитування RNA-seq Baseзвані ONT вперше були оброблені за допомогою Porechop16 (версія 0.2.4, https://github.com/rrwick/Porechop), щоб видалити послідовності адаптерів. Потім обрізані зчитування піддавали контролю якості попереднього вирівнювання та фільтрації, щоб видалити зчитування з оцінкою Phred 13 (версія 1.4, https://github.com/BrooksLabUCSC/flair). Для вирівнювання, minimap217 (версія 2.17-r941, https://github.com/lh3/minimap2) використовувався з такими параметрами: minimap2 -ax splice ./reference/ref-human-ont.mmi $i/out.pass.fq -t 10 -o $i/aln.sam. Інделі були видалені з вирівнювання, а сайти сплайсингу були уточнені за допомогою спеціальної функції FLAIR. Достовірними вважалися лише з’єднання сплайсингу, анотовані в наборі генів GENCODE v38, що підтверджувалися щонайменше трьома однозначно зіставленими зчитуваннями. Крім того, невідповідні сайти сплайсингу були виправлені шляхом відображення їх на найближчий дійсний сайт у вікні 10 нт. Цей процес гарантував, що лише точні та підтверджені події сплайсингу були включені до остаточного аналізу.
Основні статистичні дані базового секвенування нанопори довгозчитуваної РНК були підсумовані в таблиці 2. Більшість зчитувань секвенування показали високі середні показники якості (12,48) для всіх зразків (рис. 1а). Середня довжина зчитування в зразках становила приблизно 847 bp (рис. 1b). Більшість зчитувань із більшості зразків успішно зіставлено з геномом людини, але деякі зразки, зокрема AsPC-1-1 і AsPC-1-2, показали помітну частку нелюдських зчитувань (рис. 1c). Після таксономічної класифікації цих зчитувань за допомогою Kraken2 (версія 2.1.3, https://github.com/DerrickWood/kraken2) ми виявили, що більшість узгоджується з Мікоплазматіящо вказує на наявність зараження мікоплазмою, а не зараження іншими видами бактерій. Зараження мікоплазмою є загальновизнаною проблемою клітинної культури, яка може впливати на фізіологію клітини, експресію генів і результати експериментів18,19. Хоча мікоплазму не було виявлено у всіх клітинних лініях, ми визнаємо це як обмеження в уражених зразках. Важливо, що ми підтвердили, що профілі транскриптомів людини залишаються високо корельованими між біологічними репліками, що свідчить про те, що забруднення суттєво не порушило загальний транскриптомний ландшафт, зафіксований у цьому наборі даних. Тим не менш, ми рекомендуємо користувачам пам’ятати про потенційні зчитування нелюдськими засобами та рекомендуємо застосовувати стратегії фільтрації зчитування, щоб виключити їх під час аналізу виразів або подальших обчислювальних досліджень. Зчитування з усіх зразків переважно продемонструвало хорошу якість секвенування з довжиною ≥200 п.о. і показниками якості ≥7 (рис. 1d). Згодом ми відфільтрували зчитування коротші за 200 bp, зчитування з показником якості нижче 7 і зчитування, зіставлені з нелюдськими геномами. Цей крок фільтрації трохи покращив загальні середні показники якості (рис. 1e) і середню довжину читання (рис. 1f).
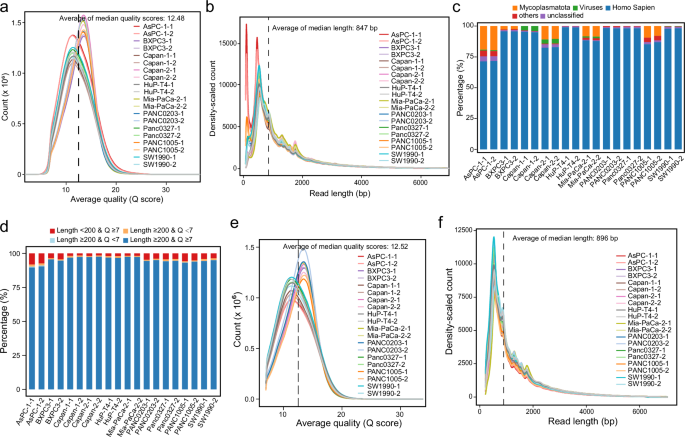
Контроль якості попереднього вирівнювання зчитувань послідовності тривалого зчитування нанопор. (a) Графік щільності, що ілюструє розподіл середніх показників якості для попередньо відфільтрованих зчитувань послідовності в кожному зразку. (b) Графік щільності, що відображає розподіл довжини зчитування для попередньо відфільтрованих зчитувань послідовності в кожному зразку. (в) Стовпчасті діаграми, що представляють відсотки читань, зіставлених на різні види. (d) Стовпчасті діаграми, що показують відсоток зчитувань із різними довжинами та оцінками якості після виключення тих, які зіставлені з нелюдськими геномами. (д) Графік щільності, що ілюструє розподіл середніх показників якості для зчитування послідовності після фільтрації в кожному зразку. (f) Графік щільності, що відображає розподіл довжини зчитування для зчитувань послідовності після фільтрації в кожному зразку.
Відфільтровані зчитування були узгоджені з еталонним геномом людини (GRCh38), що призвело до високих коефіцієнтів відображення для всіх зразків (рис. 2а). Вирівняні зчитування були кластеризовані відповідно до геномних координат (±20 bp), розташування хромосом і шаблонів сплайсингу. У кожному кластері дублікати ПЛР ідентифікували шляхом обчислення подібності попарної послідовності з використанням відстані Левенштейна, позначаючи зчитування з ≥95% ідентичністю репрезентативної послідовності як дублікати. Ця стратегія враховує специфічну для нанопор мінливість вирівнювання та помилки секвенування без ненавмисного згортання біологічно відмінних ізоформ. Дублікати ПЛР були позначені за допомогою прапора SAM (0x400), що становить приблизно 0,5% від загальної кількості зчитувань у середньому для зразків (рис. 2b). Зчитування дублікатів ПЛР були виключені з наступних кількісних аналізів транскриптів. Потім ми розрахували нормалізоване охоплення між позиціями транскрипту за допомогою інструментарію picard (версія 2.20.4-SNAPSHOT, https://github.com/broadinstitute/picard), який показав незначне зниження від кінця 3' до кінця 5' (рис. 2c). У кожному зразку приблизно 85% читань охоплювали принаймні 80% їх вирівняної довжини транскрипту (рис. 2d). Нарешті, ми оцінили рівень помилок секвенування, обчисливши пропорції невідповідностей, вставок і видалень відносно загальної кількості вирівняних баз. Отримана середня похибка для зразків становила приблизно 7% (рис. 2e). ПЛР-дублікати та помилки секвенування оцінювали за допомогою спеціальних сценаріїв. Під час аналізу внутрішнього праймування ми виявили, що приблизно 7,7% зчитувань мали гомополімерні розтягнення A (≥6 A) поблизу своїх початкових місць, що свідчить про помірний ступінь внутрішнього праймування20. Незважаючи на те, що цей артефакт не домінує, його слід враховувати під час інтерпретації меж транскрипту. Більшість зчитувань залишаються незмінними, а покриття транскриптів і кореляція між реплікатами вказують на надійність профілювання основного транскриптому.
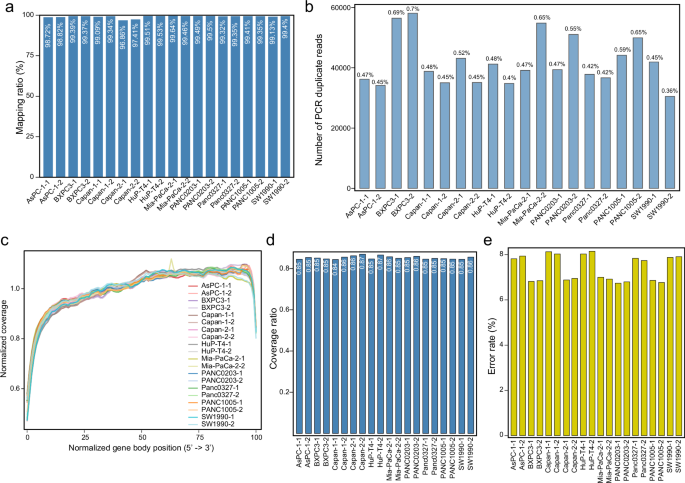
Контроль якості даних секвенування довгострокового зчитування нанопор після вирівнювання. (a) Стовпчасті діаграми, що ілюструють частку зчитувань секвенування, успішно зіставлених з еталонним геномом людини (GRCh38) для кожного зразка. (b) Стовпчасті діаграми, що відображають кількість повторних зчитувань ПЛР, ідентифікованих на зразок. (в) Лінійні діаграми, що представляють розподіл нормалізованого охоплення читання між нормалізованими позиціями транскриптів у кожному зразку, обчислення за допомогою інструментарію Picard. (d) Стовпчасті діаграми, що відображають відсоток зчитувань, що охоплюють понад 80% вирівняної довжини транскрипту в кожному зразку. (д) Стовпчасті діаграми, що показують оцінені частоти помилок секвенування, розраховані як частка невідповідностей, вставок і видалень відносно загальної кількості вирівняних баз, для кожного зразка.