Опис набору даних
Для оцінки зручності використання та універсальності трубопроводу SCSparkl ми реалізували три загальнодоступні одноклітинні набори даних RNA-SEQ, кожен з яких представляє чіткий біологічний контекст та структуру даних:
Немієлоїдні клітини миші мозок
Цей набір даних є частиною складу Tabula muris senis, який профілює одноклітинні транскриптоми у 20 тканинах від Mus musculus, що містить приблизно 100 000 клітин загалом23. Для нашого аналізу ми витягнули підмножину немієлоїдних клітин мозку, що включає 3,401 клітин з вимірюваннями експресії генів для 23 433 генів. Супровідні метадані надають детальні анотації, такі як суб тканинного походження (кора, гіпокамп, мозочок та стриатум), онтологічні класифікації клітин, ідентифікатор миші та стать.
Клітинна суміш Jurkat-293T
Цей набір даних, спочатку опублікований24містить 3,388 клітин, отриманих із суміші in vitro 1: 1 людських 293 Т та клітинних ліній миші Юркат. Набір даних був розроблений для розмежування профілів експресії, характерних для видів. Клітини, що експресують CD3D, були класифіковані як Юркат, тоді як ті, що виражають XIST, були ідентифіковані як 293T. Цей набір даних служить чітким орієнтиром для оцінки міжвидового розділення та точності кластеризації.
68 K PBMC
Для оцінки продуктивності трубопроводу на великих наборах даних та оцінки масштабованості виконання, ми використовували набір даних мононуклеарної клітини периферичної крові 68 К24 Набір даних складається з одноклітинних транскриптомічних профілів, зібраних від здорового донора, що охоплює неоднорідну популяцію імунних клітин. Цей набір даних був використаний для генерації випадкових підпрограм збільшення розмірів (від 2 К до 10 К.
Розробка трубопроводу
Для розробки ми використовували Spark Version 3.1.2, Python 3.9 та JDK версії 8.0. Apache Spark – це розподілений аналітичний двигун, зроблений для обробки великих даних. Він забезпечує основну платформу паралельної обробки для великих наборів даних25. Іскра в основі використовує абстракцію під назвою стійкі розподілені набори даних (RDD), колекція об'єктів, що розподіляється лише через кластер машин, яку можна відтворити, якщо одна з машин втрачена-толерантність12. Усі завдання запускаються паралельно за допомогою програми під назвою “Програма драйверів”, яка також підтримує високий рівень контролю над програмами та роботами, поданими виконавцям (рис. 1). Spark використовує концепцію пам’яті, тобто, вона використовує пам’ять випадкового доступу (оперативної пам’яті) машини та приносить дані, що обробляються безпосередньо до оперативної пам’яті25.
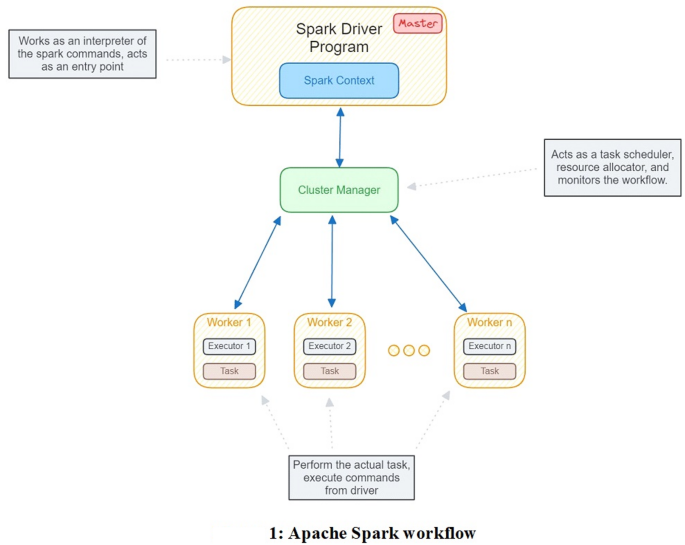
Apache Spark Workflow. Програма Spark Driver працює як майстер і як вхідна точка для всіх робочих місць Spark. Майстер подає роботу до вузлів робітників. Менеджер кластерів веде трек вузлів, а завдання розподіляються їм, кілька менеджерів кластерів – це ще один переговорник ресурсів (пряжа), кубернетти, мезо та окремі (у нашому випадку). Робітник/рабські вузли – це фактичні машини, де виконуються завдання, і вони повідомляють перед менеджером кластерів.
Трубопровід SCSparkl складається з 3 етапів, алгоритмів для всіх трьох етапів (псевдокод 1, 2 та 3) були розроблені таким чином, що нормальне обладнання для товару підтримуватиме аналіз, і можлива оптимізація іскри. Етапи мають модульний характер, що підтримує різноманітність аналізу. Для глибокого розуміння того, як працюють етапи внутрішньо, ми розробили схему потоку даних (DFD) для кожної стадії (додаткова секта 1).
Преоціація стадії-1
Для завантаження даних та фільтрації даних було розроблено два ексклюзивні модулі Spark. Файл пакету data_load.py завантажує дані як Spark Dataframe і виконує початкову попередню обробку, як видалення спеціальних символів, таких як періоди (.), Comma (,), порожні пробіли тощо, зі стовпців та замінює їх на підкреслення ('_'). Дані SCRNA-SEQ зазвичай знаходяться у широкому форматі, а Spark має можливість краще працювати з даними високого/довгого формату; Отже, для оптимізації ми переробляємо дані у високий формат за допомогою melt_spark () Функція в трубопроводі, тим самим зменшуючи стовпне навантаження та підвищуючи ефективність. Цей крок є важливим для користувачів з низькими машинами пам'яті (тобто, як мінімуми, як 4 Гб). Потім дані даних записуються як іскровий паркетний файл. Формат Apache Parquet-це формат файлу зберігання стовпців з відкритим кодом, який базується на “Алгоритмі подрібнення та складання”. Він призначений для обробки великих і складних наборів даних для більш швидких функцій агрегації. Файл, форматований паркет, стискається, що прискорює подальші аналітичні процедури у файлі. В основному ми використовуємо агрегатні функції Apache Sparks, відомі своїм швидким пошуком і менш обчислювально вичерпними/дорогими. Pseudocode-1 надає детальний контур очищення та переробку даних.

Контроль якості етапу-2
Для когерентного та точного аналізу даних SCRNA-SEQ необхідно фільтрувати клітини та гени низької якості. Ми стежили за пакетом “Scater”9Сканпі14 і Сеурат13 Для генерування різноманітних матриць контролю якості. Матриця якості обчислюється за кількістю таких параметрів, як загальні гени, виражені на клітинку, відсоток консорціуму зовнішнього РНК -контролю (ERCC), численні клітини, що експресують гени, що перевищують нуль, мітохондріальний відсоток, виявлений у кожній комірці тощо.
Якщо у нас є матриця клітини/гена IJ,
Де.
Я \ (\: \ в \: \) {комірка1, клітина2, клітина3, …, клітинап.} &.
j \ (\: \ в \: \) {Джин1, ген2, ген3, …, генм} Потім,
тоді То1 і То2 Позначають якість клітин та підсумки якості генів відповідно, можна навести наступні в Таблицях 1 та 2.
На додаток до нормальних процедур фільтрації, ми реалізували методологію фільтрації середнього рівня абсолютного відхилення (MAD) для фільтрації генів із нормалізованим значенням підрахунку, що перевищує вирішений поріг (зазвичай> 3). Pseudocode-2 окреслює загальну процедуру обчислення вимірювань якості клітин та генів, тоді як Pseudocode-3 окреслює процес фільтрації на основі цих резюме якості разом із наступними етапами нормалізації, зменшенням розмірності та кластеризацією, які детально описані в наступних розділах.

Стадія 3 Нормалізація та зменшення розмірності
Через властиву розрідженості та мінливості одноклітинних даних RNA-SEQ (SCRNA-SEQ) нормалізація є критичним кроком попередньої обробки, щоб забезпечити, щоб аналізи вниз за течією відображали справжні біологічні сигнали, а не на технічний шум. Правильна нормалізація покращує порівнянність експресії генів у клітинах і підвищує точність кластеризації, зменшення розмірності та диференціальний аналіз експресії. З data_normalize.py Модуль в рамках SCSPARKL в даний час підтримує два методи нормалізації: квантильна нормалізація (QN) та глобальна нормалізація (GN).
У QN значення експресії генів по всій клітині вперше класифікуються від найнижчого до найвищого, дані розташовані відповідно до рядків рядків. У всіх клітинах значення в одному ранзі усереднюються для формування нового еталонного розподілу. Потім ці середні значення відображаються назад до кожної комірки відповідно до початкового порядку. Цей метод допомагає стандартизувати розподіл експресії по клітинах, зберігаючи відносні відносини експресії всередині кожної клітини. Це особливо корисно для того, щоб зробити набори даних з різними масштабами виразів більш порівнянними.
Глобальна нормалізація, з іншого, регулює загальний вираз кожної клітини до фіксованої шкали (наприклад, підраховується на 10 000), а потім трансформація журналу. Цей підхід виправляє відмінності в глибині секвенування і зазвичай використовується в стандартних робочих процесах SCRNA-Seq. У цьому ми ділимо кожне значення експресії клітини на загальну кількість кількості гена. Потім отримане значення помножується на 10 000 і перетворюється журнал з додаванням псевдо-вартості 1. Математично, якщо \ (\: {X} _ {n*m} \) являє собою ненормалізована відфільтрована матриця SCRNA-Seq, а потім нормалізована матриця \ (\: {X} _ {n} \) можна дати як:
$$ \: \ start {array} {c} {x} _ {n} = \ лівий \ {\ frac {{x} _ {i, j}} {\ sum \: _ {i, j = 0}^{i \ le \: n, j \ le \: m} \ ліворуч ({x} _ {i, j} \ right)}*\ tex
(1)
$$ \: де \: x \: \ in \: {x} _ {m*n} \: і \: \ tex
і ця масштабована нормалізована матриця потім перетворюється як:
$$ \: \ start {array} {c} {x} _ {sn} = \: \ ліворуч (loglog \: \ ліворуч ({x} _ {n} \ право) +1 \: \ праворуч) \: \ end {array} $$
(2)
$$ \: де \: 1 \: є \: a \: pseudocount. \: $$
Обидві стратегії нормалізації інтегровані в іскровий трубопровід для підтримки паралельного виконання та ефективної обробки масштабних наборів даних.
Зменшення розмірності
Зниження розмірності є ключовим кроком в аналізі SCRNA-Seq, спрямованого на проектування високовимірних даних експресії генів у нижньовимірний простір, зберігаючи основну структуру та взаємозв'язки між клітинами26. Через високу розмірність та розрідженість даних SCRNA-seq, звичайні методи аналізу можуть впливати на «прокляття розмірності»-явище, де відстань між точками даних стає менш значущим у високомірних просторах27.
У цьому дослідженні ми використовували три широко використовувані методи зменшення розмірності: аналіз основних компонентів (PCA), Т-розподілене стохастичне вбудовування сусіда (T-SNE) та рівномірне наближення та проекцію (UMAP). Серед них PCA реалізується за допомогою бібліотеки MLLIB Spark, що дозволяє розподілити та паралельні обчислення через вузли. Ця інтеграція узгоджується з проектними цілями SCSparkl для підтримки масштабованого, високопропускного аналізу.
Навпаки, UMAP та T-SNE в даний час реалізуються за допомогою стандартних бібліотек Python та працюють поза двигуном виконання іскри. Тим не менш, ці методи читають вхід безпосередньо з проміжних файлів Apache Parquet, що виграють від ефективного вводу/виводу та сумісності з іскровими випусками. Незважаючи на те, що ці кроки не повністю паралельовані в іскрі, їх інтеграція в трубопровід залишається безшовною і підтримує візуалізацію та кластеризацію робочих процесів.
Кластеризація та подальший аналіз за течією
Основна перевага одноклітинної секвенування РНК (SCRNA-Seq) полягає в його здатності ідентифікувати як відомі, так і нові популяції клітин за допомогою непідконтрольної кластеризації28. По мірі того, як технології одноклітинних профілювання продовжують розвиватися, поліпшення ефективності захоплення дозволило послідовності мільйонів окремих клітин, що спричиняє все більш великі та складні набори даних. Ця шкала накладає значні обчислювальні вимоги до алгоритмів кластеризації, що використовуються для виявлення та класифікації типу клітин29.
Щоб вирішити це, SCSparkl інтегрує алгоритм кластеризації k-means, який розділяє набір даних N спостережень k кластери (k ≤ n) шляхом мінімізації дисперсії в межах кластера. Алгоритм працює ітеративно, спочатку шляхом випадкового вибору k -центроїд, а потім присвоюючи кожну точку даних найближчому центроїд. Цей процес триває до конвергенції, що призводить до компактних та добре розбитого кластерів.
Для підтримки усвідомленого відбору kТрубопровід включає як метод лікоть, так і аналіз балів силуетів, що дозволяє користувачам оцінювати стабільність та розділення кластеризації. Після визначення кластерів диференціальний аналіз експресії генів (DGE) проводиться для виявлення генів, які найбільш виразно експресуються між групами. Для цього ми застосовуємо двопробний t-тест у кластерах і класифікуємо 10 значущих генів на основі їх p-значень, забезпечуючи інтерпретаційний набір маркерних генів для біологічної перевірки нижче за течією.
